1.4 测试与表征
FTIR:采用Bruker公司Vector-22型红外光 谱仪,溴化钾压片。
SEM:在扫描电镜载物台上贴上导电双面胶, 撒少许样品后吹去多余粉末,表面喷金后抽真空观 察,电压加速度为20 kV。
粒度分析:将样品在蒸馏水中超声分散20 min 后加入PS比色皿中,利用Brookhaven公司Zeta-plus激光粒度分布仪测试其粒径及其分布。
2 结果与讨论
2.1 2E4MI-g-BGE的合成
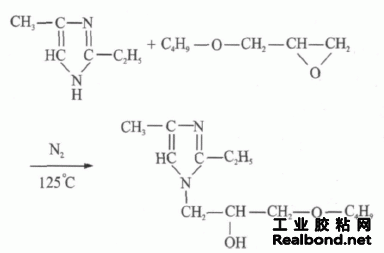
为了避免在包囊过程中部分芯材溶入水相,本实验选择了在2-乙基-4-甲基咪唑分子接枝上极性弱的基团,即在仲胺位置上引入庞大的脂肪烃侧基基团,改性剂为丁基缩水甘油醚,改性产物为水不 溶性,并可以作为环氧树脂的固化剂[6]。合成反应过程如下:
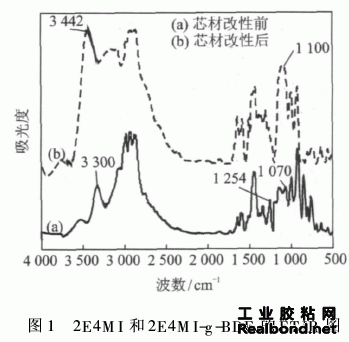
通过红外光谱图(见图1),三元环醚基在1 254 cm-1、950~810 cm-1及840~750 cm-1处有吸收峰存在,这3个峰可以作为三元环醚存在的标志,仲胺中的N—H键的伸缩振动峰在3300 cm-1左右, 吸收较弱,而1000~1 110 cm-1之间是C—N—C 键的吸收峰,这些是芯材改性前曲线(a)的特征吸 收峰位。芯材改性后的曲线(b)上,三元环醚基峰 位不再明显存在,仲醇基团中的C—O键的峰位在1100 cm-1,—OH键的伸缩振动在3 200~3 500 cm-1附近,变形振动在1 350~1 260 cm-1之间,峰比较弱,带形较宽。3300 cm-1处的N—H键消失。红外分析可以看出,反应后三元环醚基转变为—OH,而仲胺基也转变成叔胺基,说明丁基缩水甘油醚成功接枝在2-乙基-4-甲基咪唑上。水溶性实验证明改性产物不溶于水。
2.2 微胶囊的制备
2.2.1 分散剂浓度的影响
在微胶囊的制备中,分散剂起着十分重要的作用。在溶剂蒸发法中,液滴的形成靠外力搅拌作用 的同时,还要有表面活性剂的乳化作用,这个过程 时间比较长,需要乳化体系很稳定,因此,选择合适 的水相分散剂对于成功包囊很关键[7],而且不同的 分散剂浓度对于微胶囊的粒径有一定的影响[8-9]。 此外,由于芯材为液状物,它的界面张力是一个恒定值,与壁材的界面张力差值越大,囊芯微胶囊的 包覆效率就越高,合适的表面活性剂可以增大芯材 的界面张力使其更容易被包囊。本文选择了常用的 聚乙烯醇和非离子聚氧乙烯类表面活性剂做了对 比实验。在相同的条件下,使用聚氧乙烯类乳化剂 在蒸发过程中粒子团聚不能形成微胶囊,而聚乙烯 醇可以成功包囊,使用不同含量聚乙烯醇(重量百 分数)的微胶囊的扫描照片如图2所示。
从图2可以看出,不同分散剂浓度下得到的微胶囊表面形貌不同,当浓度为2.5%时胶囊为椭球 形,并且表面致密性很差;当浓度为5%,8%时可得到很规则球状胶囊。从图3的粒径分布图可以看到,3种浓度下的粒径均为正态分布,浓度为5%时制得的微胶囊粒径分布0.80~7.37μm,浓度为 8%时微胶囊粒径分布0.58~1.98μm,浓度为 10%时微胶囊粒径分布0.51~1.62μm。为了得到小粒径窄分布的微胶囊,同时考虑到聚乙烯醇不能溶于环氧树脂中,过量加入时不利于产品的后处 理,所以实验中选用聚乙烯醇浓度为8%。